Motivation
As with many folks during the long dark winter months, I needed something to keep me busy. As part of my PhD, I have had the opportunity to sequence a few genomes. Each time it has taken weeks or months between DNA and data. To circumnavigate this, I trialled Oxford Nanopore’s miniature genome sequencer which plugs right into your laptop and gives you super long reads!
In the Matthew’s lab, we work on a broad diversity of the protozoan genus, Trypanosoma. T. melophagium is a fascinating example of a Trypanosoma which, in contrast to the more infamous T. brucei, is non-pathogenic. T. melophagium can be found in temperate regions where pesticide has not been widely used, such as the barren and desolate isles of St. Kilda, off of the Scottish coast. T. melophagium is transmitted from host to host (Ovinus) via the sheep ked. It is supposedly closely related to another Trypanosoma, T. theileri which can be found in similar climates but is transmitted between bovids via tabanids.
Interest has grown around these trypanosomes as researchers in Edinburgh, some from the Matthews lab, have begun a study using T. melophagium and T. theileri as a vaccine vechicle.
T. theileri has had its genome sequenced which revealed an alternative survival technique in the mammalian bloodstream, compared to T. brucei.
T. melophagium represented an exciting opportunity to test out the minION sequencer whilst providing a fascinating study to identify the genomic basis of host/ vector specificity in two closely related Trypanosomes.
Data acquisition
High molecular weight DNA was extracted and loaded onto the minION. The flow cell happily chugged away for ~10 hours and produced ~30x coverage of the genome. It wasn’t a spectacular run but I was happy with my first solo minION run! 30x coverage would do fine for creating a scaffold assembly. Especially with a few miniature whale reads (max read length = 249,891). I also generated ~100x coverage with BGI’s DNBseq.

Initial data assessment
This project also provided my first opportunity to supervise a project. I entailed the help of a final year honours student who started as a near novice to the command line and bioinformatics. Within a couple of weeks, she was swiftly navigating the command line like the high seas and it was time to sink her teeth into the data.
Firstly, we wanted to check the predicted genome size and heterozygosity with an assembly independent tool. We selected a k-mer counting based approach of Jellyfish/ Genomscope. We chucked the T. melophagium and T. theileri short reads at it and to our surprise, the supposedly closely related T. theileri genome was 5.4Mb larger than T. melophagium! A large proportion of this variation was precited to arise from repeats in T. theileri. Looking at phylogenetic trees, T. grayi is the next closest relative to T. melophagium and has a genome ~21Mb. Therefore, unless both T. theileri and T. grayi separately underwent genome loss, T. theileri appears to have expanded its genome.
With our interest truly peaked, we proceeded to genome assembly.
Genome assembly
After scanning recent papers which assembled minION data with short reads to polish, we narrowed down our pipeline to a combination of redbean to generate a raw assembly and a combination of racon, medaka and pilon which used the long and/or and short reads as inputs. After each round of polishing BUSCO and scaffold stats were calculated. Our final round of polishing produced a 66 contig assembly which hit a BUSCO score of 100% complete and an N50 of 505,851 🎉
Hello host/vector specific genes, my old friends
With a decent draft T. melophagium genome to add to the ~40 TriTrypdDB genomes, we began clustering orthologous proteins with Orthofinder.
Where has that 5.4Mb come from!! Some tantalisingly low hanging fruit jumped out, such as the absence of a surface protein family in T. melophagium and hugely expanded surface protein families in T. theileri.
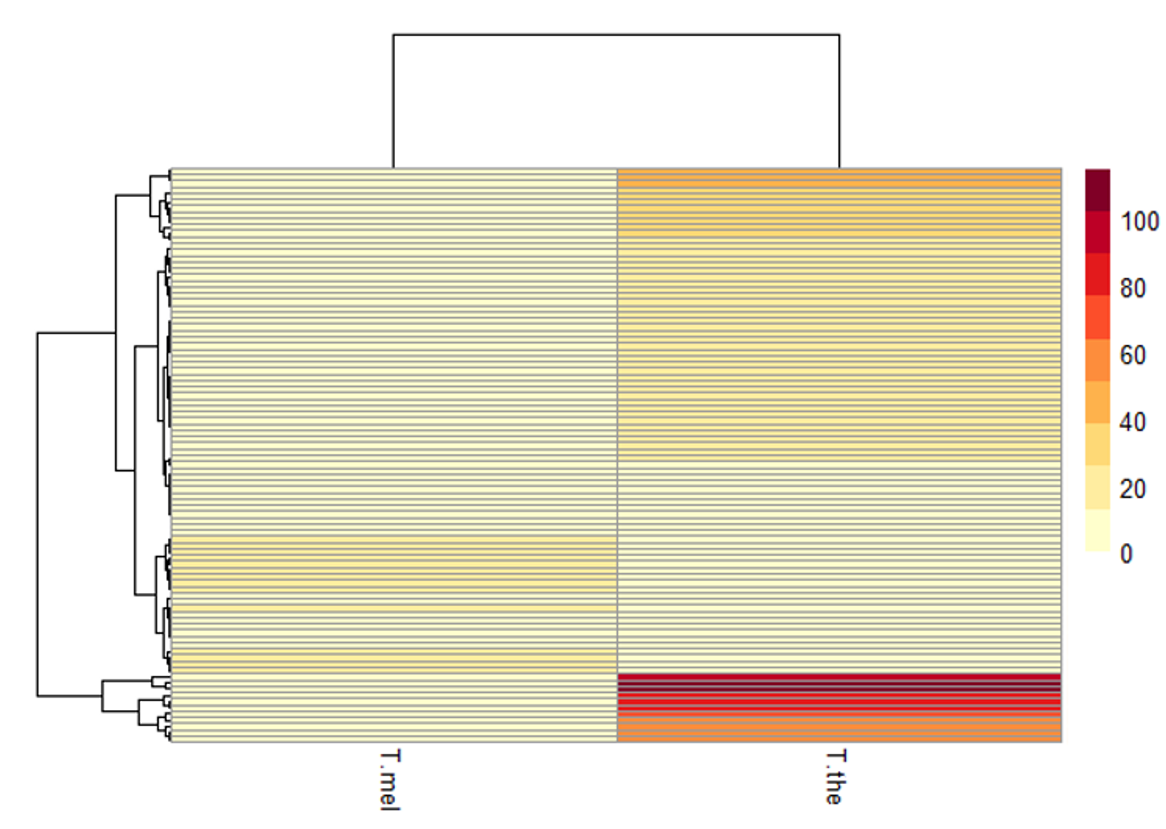
And that’s all the progress so far, stay tuned!